
Conhecendo a mucopolissacaridose tipo II
Conhecida também como Síndrome de Hunter, a mucopolissacaridose tipo II (MPS II) é um distúrbio genético raro que afeta quase que exclusivamente indivíduos do sexo masculino em diferentes partes do corpo.1
Ao surgir entre os 2 e 4 anos de idade, o problema geralmente se manifesta na infância com sintomas diferentes em cada paciente1,2, variando entre:
alterações no formato da face;3
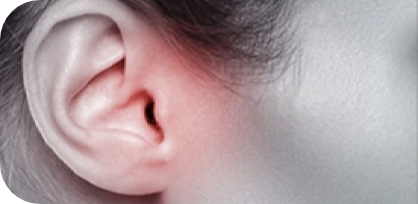
infecções de ouvido frequentes;3,4
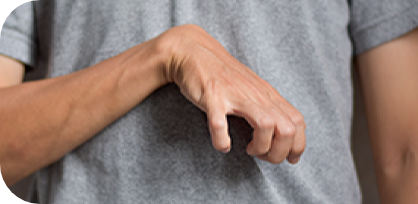
mãos em garra;3,4
infecções respiratórias frequentes;3,4

hérnia umbilical;3-4

abdômen saliente;3-4
baixa estatura;3-4

ponta dos pés;3-4

Antigamente, os açúcares glicosaminoglicanos eram conhecidos como mucopolissacarídeos e, por isso, essa doença de acúmulo recebeu então o nome de Mucopolissacaridose.6
A MPS II acontece quando as enzimas não conseguem quebrar adequadamente certas moléculas. Diante da deficiência ou ausência total da enzima iduronato-2-sulfatase (I2S), os glicosaminoglicanos (GAGs) não são quebrados e se acumulam, impedindo que o corpo funcione de forma adequada.1,2

Por afetar diversas partes do corpo, é considerada uma doença multissistêmica e pode ser considerada neuronopática ou não neuronopática, diferenciando-se no tempo da manifestação dos sintomas, progressão, deficiência cognitiva e regressão neurológica.1,2,7

Seu diagnóstico pode ser feito em laboratório ou no momento do nascimento, medindo a atividade da enzima I2S no tecido placentário ou no líquido aminiótico.2,7
Para a convivência com a doença, são necessárias certas adaptações em casa, na alimentação e outras partes da vida do paciente.8 Por isso, nos primeiros sinais, busque um especialista.
Conheça mais sobre a mucopolissacaridose tipo II, clique aqui!
Referências: 1. Scarpa M. Mucopolysaccharidosis Type II. Gene Rev. 2007. Disponível em: <https://pubmed.ncbi.nlm.nih.gov/20301451>. Acesso em março de 2024. 2. Dʹavanzo F, Rigon L, Zanetti A, Tomanin R. Mucopolysaccharidosis type II: One hundred years of research, diagnosis, and treatment. Int J Mol Sci 2020; 21. DOI:10.3390/ijms21041258. 3. Martin R et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 121, 377–86 (2008). 4. Wraith JE et al. “Initial report from the Hunter Outcome Survey.” Genetics in medicine: official journal of the American College of Medical Genetics vol. 10,7 (2008): 508-16. 5. Link B et al. “Orthopedic manifestations in patients with mucopolysaccharidosis type II (Hunter syndrome) enrolled in the Hunter Outcome Survey.” Orthopedic reviews vol. 2,2 (2010): e16. 6. Iwabe C, Frezzato RC, Nogueira AL. Evolução motora de paciente com mucopolissacaridose tipo 1. Rev. paul. pediatr. 28 (3) • Set 2010. 7. Protocolo Clínico e Diretrizes Terapêuticas Mucopolissacaridose do Tipo II. Portaria Conjunta Nº 16, de 24 de maio de 2018. Disponível em: <https://www.gov.br/saude/pt br/assuntos/pcdt/arquivos/2018/pcdt_mps-ii.pdf>. Acesso em março de 2024. 8. Hunter Patients, Takeda. Disponível em: https://www.hunterpatients.com/home-life/. Acesso em março de 2024.